Sir,
Pulmonary thrombosis is a common event in COVID -19 pneumonia and can be fatal as it leads to pulmonary thromboembolic episodes.
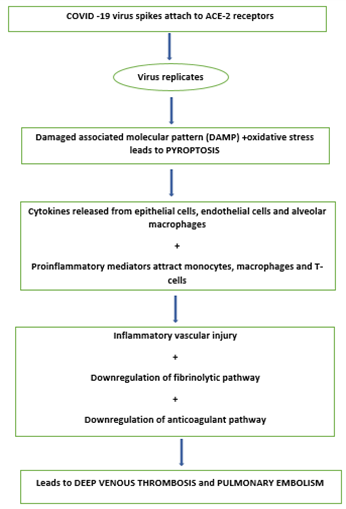
The virus has a prediliction for respiratory epithelium and enters by binding to angiotensin-converting enzyme 2 (ACE-2) found on alveolar epithelium and endothelium.
Activation of endothelial cells is the driver for causing thromboembolic episodes. Pyroptosis is the initial he immune dysregulation occurring in COVID-19 infection in which rapid viral replication occurs leading to massive release of inflammatory mediators. Although many inflammatory processes can influence D-dimer levels but in covid -19 infection it is one of the most consistent finding. An elevated D-dimer continues to be one of the most consistent markers of poor outcome.
D-Dimer levels help in categorising patients as critically and non-critically ill and thereby help in better patient management.
The early pathogenic event includes a widespread endothelitis affecting multiple organ systems with presence of viral inclusion bodies within endothelial cells accompanied by apoptosis, inflammatory cell infiltration and microvascular thrombosis. Systemic inflammation is characterised by elevated levels of C-reactive protein, fibrinogen and cytokines such as interleukin-6. Although the exact mechanisms of COVID-19-induced thrombosis have not been elucidated, some well-described mechanisms associated with infection/inflammation are likely to be relevant in initiating the event. (Figure 1)
These include:
Increased production of tissue factor and amplification of the coagulation cascade, resulting in increased production of thrombin and fibrin.1
Thromboelastography in patients with COVID-19 suggest clot formation is extremely rapid and also resistant to breakdown. This, in turn, is likely to due to reduced fibrinolysis as a result of increased production of plasminogen activator inhibitor-1 (PAI-1).
Deposition of components of the complement system, such as C5b-9, in damaged vessels of COVID-19 patients indicate that this may be another important pro-thrombotic mechanism.
Neutrophil extracellular traps (NETs) have also been observed in vessels in autopsy specimens of patients with COVID-19 .These are associated with high circulating levels of cell-free DNA and histones, which in turn can activate pro-thrombotic pathways leading to increased thrombin production.2
In addition to ACE-2-mediated SARS-CoV-2 viral entry, other recent reports of affinity of the SARS-CoV-2 spike protein and CD147, a membrane glycoprotein and extracellular matrix metalloproteinase inducer expressed on a variety of haematopoietic cell lines, suggest another potentially novel mechanism of thrombosis and inflammation in the arterial and venous circulations.
Profound hypoxaemia causes activation of hypoxia inducible factors that activate cytokines, tissue factor and PAI-1 leading to vasoconstriction, inflammation and thrombosis.
The high rate of pulmonary thrombosis in COVID-19 lies in the confluence of three processes: firstly, the intense endothelial inflammation leading to “in situ” thrombosis, including microvascular thrombosis; secondly, altered pulmonary blood flow in response to the parenchymal process, disturbing Virchow's triad within the lung; and thirdly, classical Deep venous thrombosis to Pulmonary embolism transition.
There should be a high suspicion for thrombotic complications and low threshold for definitive imaging, where possible. The intimate association between inflammation and thrombosis would suggest an anti-inflammatory/anti-viral therapeutic approach should be considered in parallel to anticoagulation.3
Thromboelastography and platelet function tests reveal that patients with COVID-19 have activated platelets suggesting that antiplatelet therapy might have some therapeutic role to play.